Accelerating Multi-Omics Analysis with High-Performance Computing
Wednesday, June 24, 2026 3:45 PM to 5:15 PM · 1 hr. 30 min. (Europe/Berlin)
Foyer D-G - 2nd Floor
Women in HPC Poster
AI Applications powered by HPC TechnologiesBioinformatics and Life SciencesDevelopment of HPC SkillsPerformance MeasurementSystem and Performance Monitoring
Information
Poster is on display and will be presented at the poster pitch session.
High performance computing (HPC) has become essential for analysing increasingly large and complex multi omics datasets. Modern biomedical research frequently integrates diverse molecular layers—including genomics, epigenomics, proteomics, metabolomics, and phenomics—to achieve a more comprehensive understanding of disease mechanisms. However, the scale, heterogeneity, and computational intensity of such analyses exceed the capabilities of standard desktop environments. This work demonstrates how HPC infrastructures accelerate multi omics workflows, improve computational stability, and support reproducible large scale analyses using data from the Northern Ireland Cohort for the Longitudinal Study of Ageing (NICOLA).
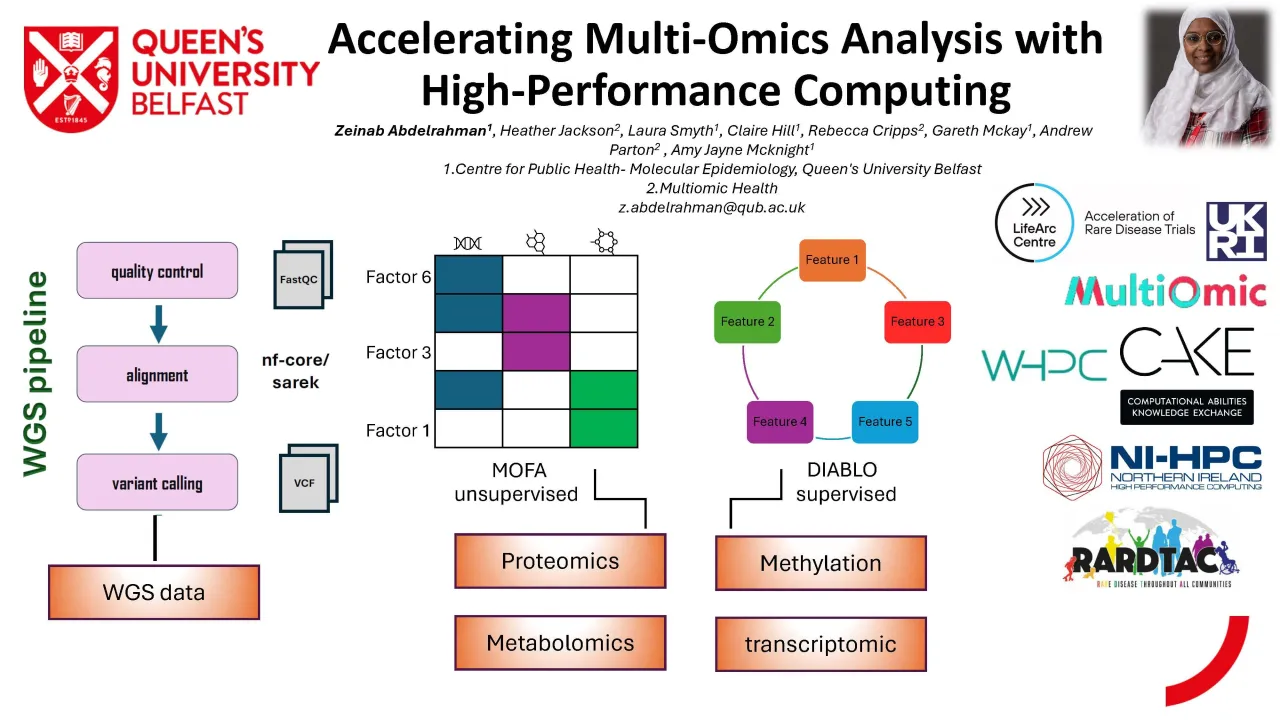
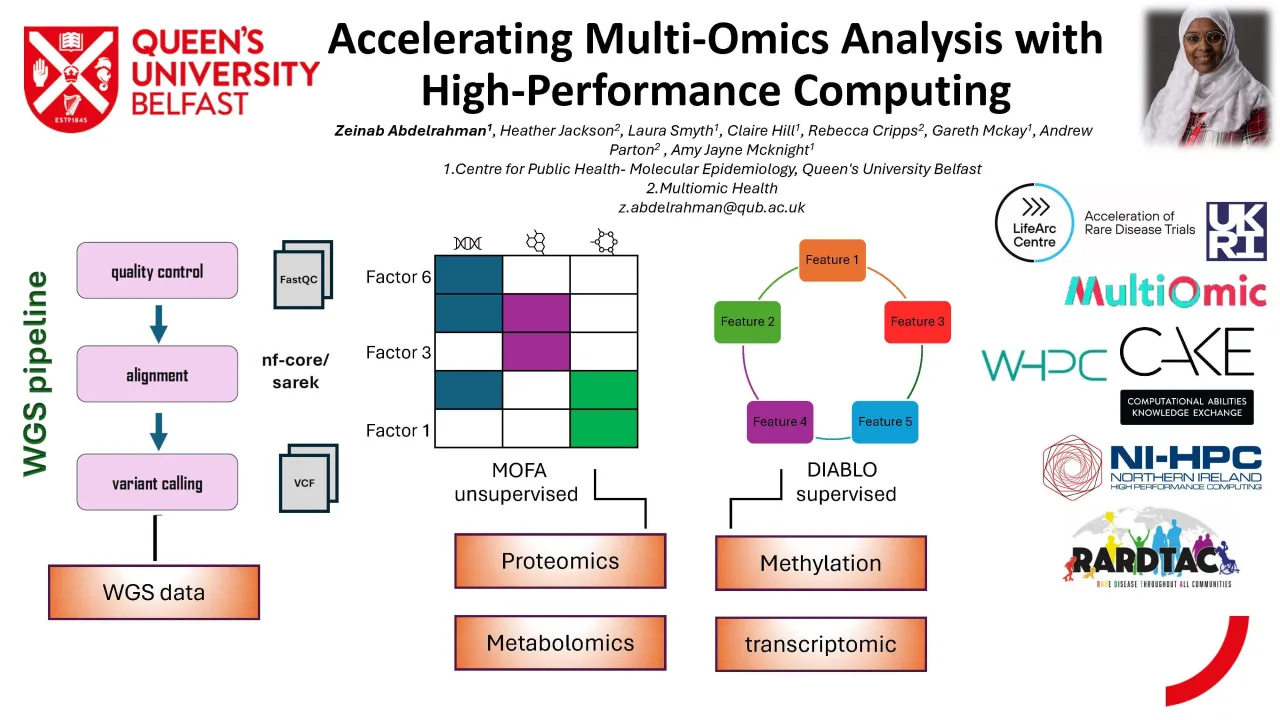
Whole Genome Sequencing (WGS) presents one of the most computationally demanding components of multi omics pipelines. The processing of WGS data requires sequential steps involving quality control, alignment to a reference genome, variant calling, and annotation. Using the FastQC module and the nf-core/sarek pipeline on an HPC cluster, we were able to efficiently process ~1 TB of sequencing data across 146 samples. The parallelisation and memory optimisation achievable on HPC reduced turnaround times substantially. Workflows that typically require multiple days on a standard desktop—often resulting in system crashes or memory failures—were completed within 24 hours on HPC. This demonstrates the critical role of HPC in delivering scalable, reliable WGS analysis for medium to large cohorts.
Beyond sequencing, multi omics integration poses its own computational challenges. For the NICOLA cohort, we incorporated several datasets of varying sizes and complexity: epigenetic profiles (~900,000 CpG sites, ~2 GB), Biocrates metabolomics (~1019 metabolites), Nightingale NMR metabolomics (~247 metabolites), and OLINK proteomics (~5000 proteins, ~28 MB). We employed Multi Omics Factor Analysis (MOFA) to uncover shared latent structures and cross platform molecular signatures. MOFA’s computational demands scale rapidly with increasing sample size and feature numbers, often exceeding the memory capacity of typical desktop machines. Running MOFA on the HPC cluster reduced runtime from 4–6 hours to approximately 3 minutes and eliminated common issues such as crashes during matrix factorisation, highlighting the importance of HPC in enabling stable high dimensional modelling.
Overall, HPC provided four major advantages for this research: (1) accelerated analysis, enabling rapid turnaround for both sequencing and integrative modelling; (2) stability, preventing crashes during memory intensive steps; (3) reproducibility, supported through structured job scripts and shared environment modules; and (4) efficient resource utilisation, ensuring optimal balancing of CPU and memory loads across the cluster. Together, these benefits significantly enhance the feasibility, robustness, and scalability of advanced multi omics epidemiological analyses.
This work illustrates how HPC transforms the analytical capacity of biomedical research by overcoming computational bottlenecks and enabling the integration of diverse biomolecular datasets. As multi omics continues to expand in complexity and scale, HPC will remain an indispensable tool for unlocking deeper biological insights and supporting reproducible, large cohort molecular epidemiology.
High performance computing (HPC) has become essential for analysing increasingly large and complex multi omics datasets. Modern biomedical research frequently integrates diverse molecular layers—including genomics, epigenomics, proteomics, metabolomics, and phenomics—to achieve a more comprehensive understanding of disease mechanisms. However, the scale, heterogeneity, and computational intensity of such analyses exceed the capabilities of standard desktop environments. This work demonstrates how HPC infrastructures accelerate multi omics workflows, improve computational stability, and support reproducible large scale analyses using data from the Northern Ireland Cohort for the Longitudinal Study of Ageing (NICOLA).
Whole Genome Sequencing (WGS) presents one of the most computationally demanding components of multi omics pipelines. The processing of WGS data requires sequential steps involving quality control, alignment to a reference genome, variant calling, and annotation. Using the FastQC module and the nf-core/sarek pipeline on an HPC cluster, we were able to efficiently process ~1 TB of sequencing data across 146 samples. The parallelisation and memory optimisation achievable on HPC reduced turnaround times substantially. Workflows that typically require multiple days on a standard desktop—often resulting in system crashes or memory failures—were completed within 24 hours on HPC. This demonstrates the critical role of HPC in delivering scalable, reliable WGS analysis for medium to large cohorts.
Beyond sequencing, multi omics integration poses its own computational challenges. For the NICOLA cohort, we incorporated several datasets of varying sizes and complexity: epigenetic profiles (~900,000 CpG sites, ~2 GB), Biocrates metabolomics (~1019 metabolites), Nightingale NMR metabolomics (~247 metabolites), and OLINK proteomics (~5000 proteins, ~28 MB). We employed Multi Omics Factor Analysis (MOFA) to uncover shared latent structures and cross platform molecular signatures. MOFA’s computational demands scale rapidly with increasing sample size and feature numbers, often exceeding the memory capacity of typical desktop machines. Running MOFA on the HPC cluster reduced runtime from 4–6 hours to approximately 3 minutes and eliminated common issues such as crashes during matrix factorisation, highlighting the importance of HPC in enabling stable high dimensional modelling.
Overall, HPC provided four major advantages for this research: (1) accelerated analysis, enabling rapid turnaround for both sequencing and integrative modelling; (2) stability, preventing crashes during memory intensive steps; (3) reproducibility, supported through structured job scripts and shared environment modules; and (4) efficient resource utilisation, ensuring optimal balancing of CPU and memory loads across the cluster. Together, these benefits significantly enhance the feasibility, robustness, and scalability of advanced multi omics epidemiological analyses.
This work illustrates how HPC transforms the analytical capacity of biomedical research by overcoming computational bottlenecks and enabling the integration of diverse biomolecular datasets. As multi omics continues to expand in complexity and scale, HPC will remain an indispensable tool for unlocking deeper biological insights and supporting reproducible, large cohort molecular epidemiology.
Format
on-demandon-site
